beta talasemia
TRANSCRIPT

BETA TALASEMIA
2011

Introducción
Conjunto de desordenes sanguíneos hereditarios, caracterizado por anomalías en la síntesis de las cadenas beta de las hemoglobinas.
Su consecuencia es un rango fenotípico que varía de la anemia severa a individuos clínicamente asintomáticos
La incidencia total anual de individuos sintomáticos es de 1 cada 100.000 individuos en el mundo.

Introducción
Las beta talasemias son causadas por mutaciones puntuales, o, más raramente, por deleciones en el gen de las cadenas beta en el cromosoma 11, de la síntesis de dichas cadenas esta reducida (beta+) o ausente (beta0).
La transmisión es autosómica recesiva, sin embargo, mutantes dominantes han sido incluso notificadas.

Introducción
la enfermedad cardíaca constituye la principal causa de muerte en pacientes con altos niveles de hierro.
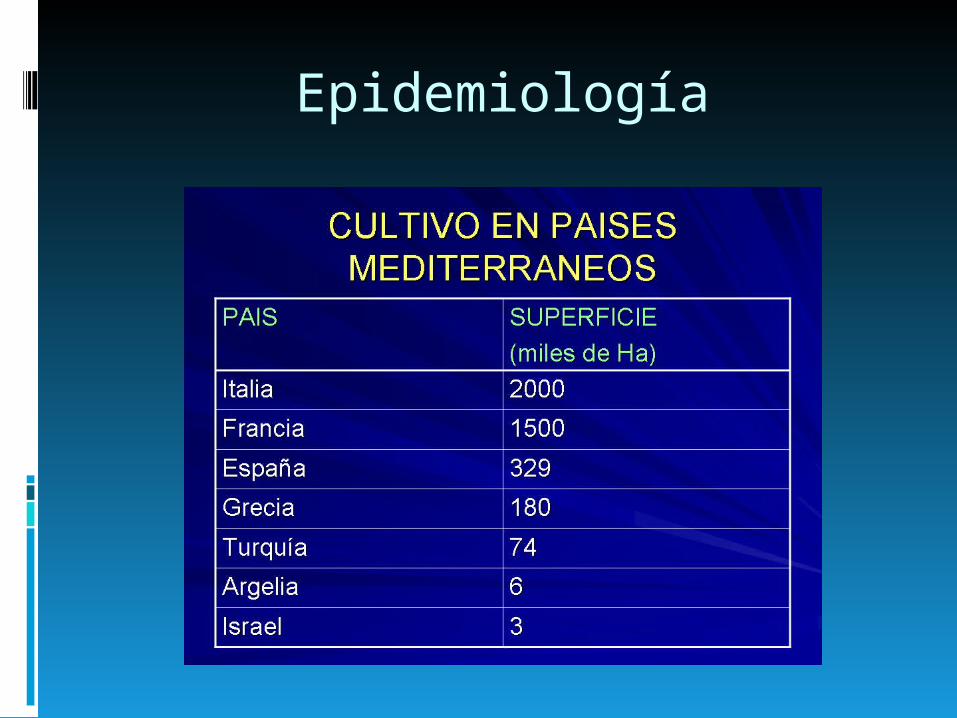
Epidemiología

Epidemiología
Salud | 8 de mayo “Día Internacional de la Talasemia”
Alrededor del 2% de la población argentina padece Talasemia y lo ignora
Sábado, 07 de Mayo de 2011 - 12:30
Entre uno y dos de cada 100 argentinos padece talasemia -o anemia del Mediterráneo- aunque la mayoría lo ignora por no manifestar síntomas, pero puede transmitirla a su descendencia en un nivel de mayor seriedad, informaron especialistas, con motivo de celebrarse mañana el Día Internacional de la Talasemia.
La Fundación Argentina de Talasemia (FUNDATAL), se sumó a más de 90 países que realizarán actividades respectivas, en su caso mediante la difusión para que la gente compruebe si padece esta afección y, en caso de ser así, tome medidas preventivas respecto de su descendencia.
Debido a la alta inmigración proveniente de países del Mediterráneo, en especial España, Italia, Líbano, Siria y Grecia, la afección alcanza ‘en distintos grados, a un amplio espectro de nuestra población, que en su mayoría desconoce padecerla‘, dijo a Télam una fuente de FUNDATAL.

Tipos de β-talasemias β-talasemias Talasemia mayor Talasemia intermedia Talasemia menor Persistencia hereditaria de la
hemoglobina fetal β-talasemias asociadas a hemoglobina HbC/ β-talasemia HbE/ β-talasemia HbS/ β-talasemia

Talasemia mayor
Genotipo: homocigota βº. Los individuos con talasemia mayor
usualmente presentan dentro de los 2 primeros años de vida una severa anemia
En pacientes no tratados o pobremente transfundidos:
Retardo en el crecimiento hepatoesplenomegalia desarrollo de masas por
hematopoyesis extramedular cambios esqueléticos
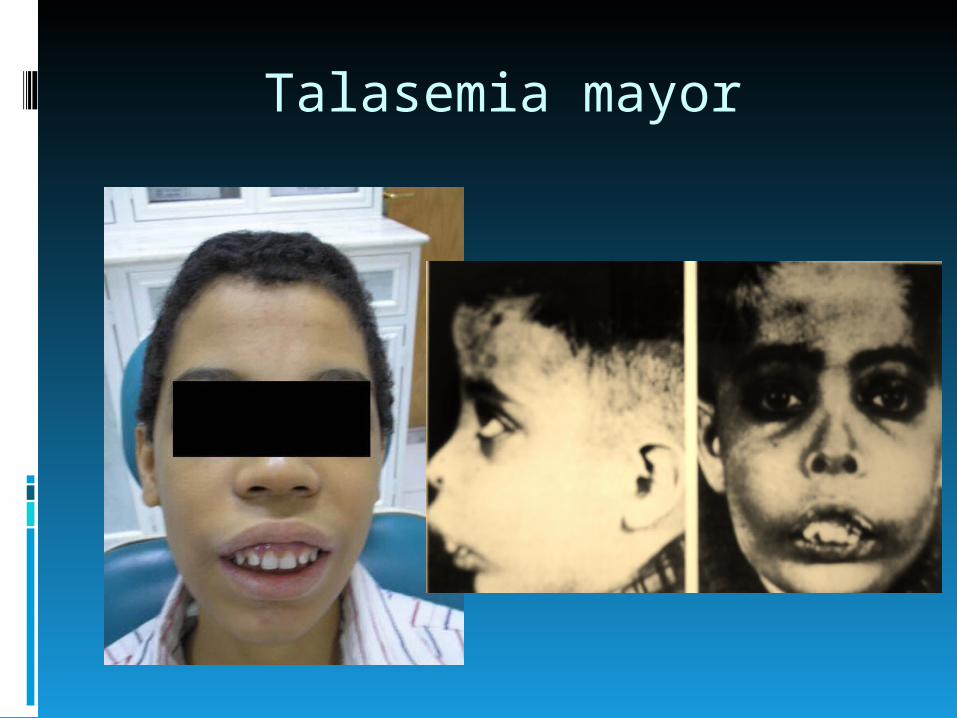
Talasemia mayor

Talasemia mayor
Requieren regularmente transfusiones de GR.
Este método trae consigo como consecuencia la sobrecarga de hierro.
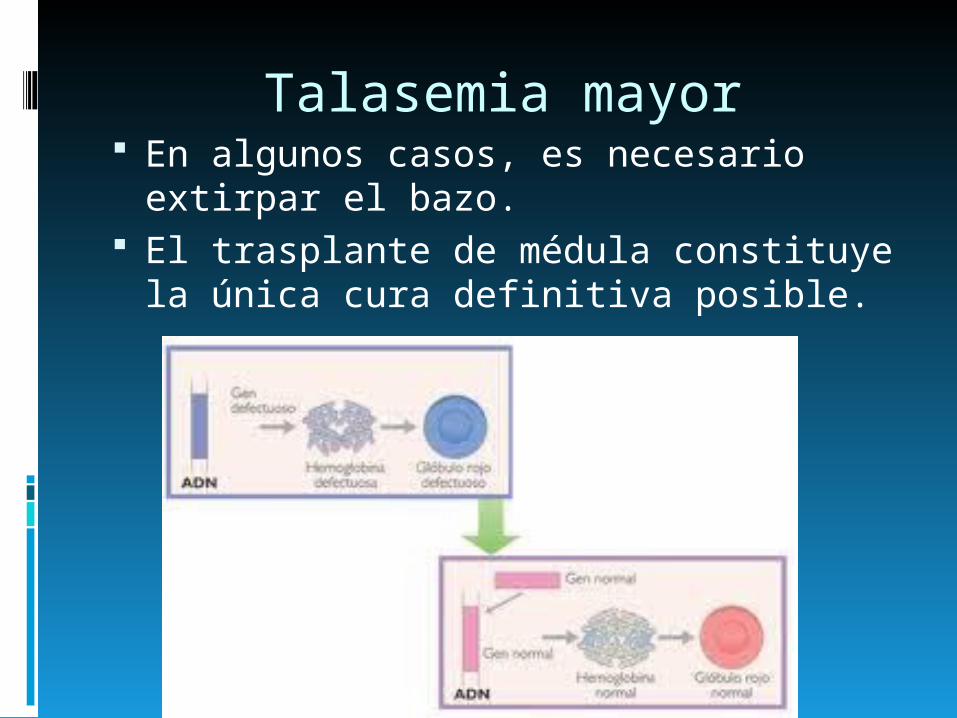
Talasemia mayor En algunos casos, es necesario
extirpar el bazo. El trasplante de médula constituye la
única cura definitiva posible.

Talasemia intermedia
La talasemia intermedia comienza a manifestarse más tarde, con anemia moderada

Talasemia intermedia
Los principales síntomas clínicos constituyen la hipertrofia de la médula eritrocitaria con hematopoyesis medular y extramedular con sus respectivas complicaciones: Deformaciones en los huesos cambios faciales típicos Cálculos biliares Ulceras en las piernas Mayor predisposición a trombosis.

Talasemia intermedia
Tratamientos:
Pueden requerir esplenoctomía Suplementación con ácido fólico Tratamiento de las masas eritropoyética
extramedulares Úlceras en las piernas

Talasemia menor
La talasemia menor es clínicamente asintomática, pero algunos pacientes suelen presentar anemia leve.

β-talasemia asociada con otras anomalías Hb

La Interacción de Hb E y β-talasemia
Leve: nivel de Hb entre 9 y 12 g/dl (asintomática)
Moderadamente severa: nivel de Hb entre 6 y 7 g/dl (similar a talasemia intermedia)
Severa nivel de Hb 4,5 g/dl (síntomas y tratamiento de talasemia mayor)

Interacción de Hb C con β-talasemia
Vida asintomática, pero en ocasiones puede tener manifestaciones clínicas:
Anemia Agrandamiento del bazo Microcitosis Hipocromía Cristales de Hb C Las transfusiones de sangre rara vez
son necesarias.

Asociación de la persistencia hereditaria de la Hb F con β-
talasemia
Aminora las manifestaciones clínicas que varían desde asintomáticas hasta talasemia intermedia.

Etiología
200 mutaciones hasta el momento Mutaciones puntuales en el gen de la
β- globina en el cromosoma 11. las deleciones del gen de globina β
son poco comunes. La mutación causa reducción o
ausencia de producción de la cadena.

Tipos de mutaciones puntuales Mutaciones transcripcionales Mutaciones en la modificación del
ARN (cap 5´) Mutaciones en la modificación del
ARN (clivaje y poliadenilación) Mutación en la modificación del ARN
(corte y empalme) Mutaciones traduccionales

Tipos de mutaciones puntuales
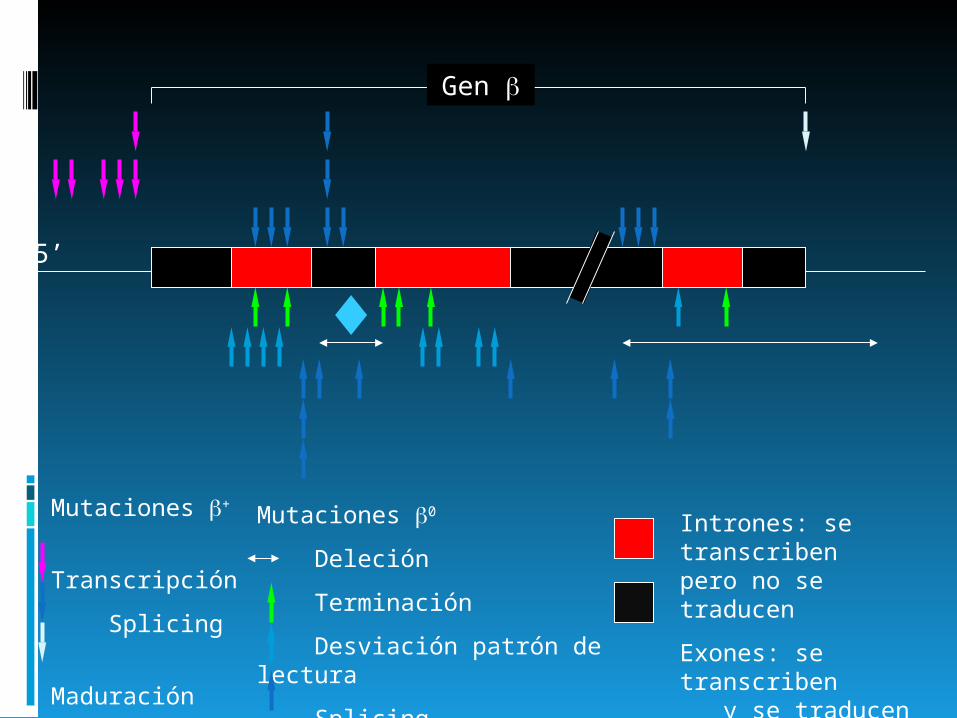
5’
Gen
Intrones: se transcriben pero no se traducen
Exones: se transcriben y se traducen
Mutaciones +
Transcripción
Splicing
Maduración
Mutaciones 0
Deleción
Terminación
Desviación patrón de lectura
Splicing

Fisiopatología
Eritropoyesis ineficaz debido a la destrucción de precursores eritroides en la MO.
Hemolisis resultante de la destrucción de lo GR circulantes que contienen inclusiones de cad α
Aumento de eritropoyetina debido a la hipoxia resultante de la ineficacia de los GR.
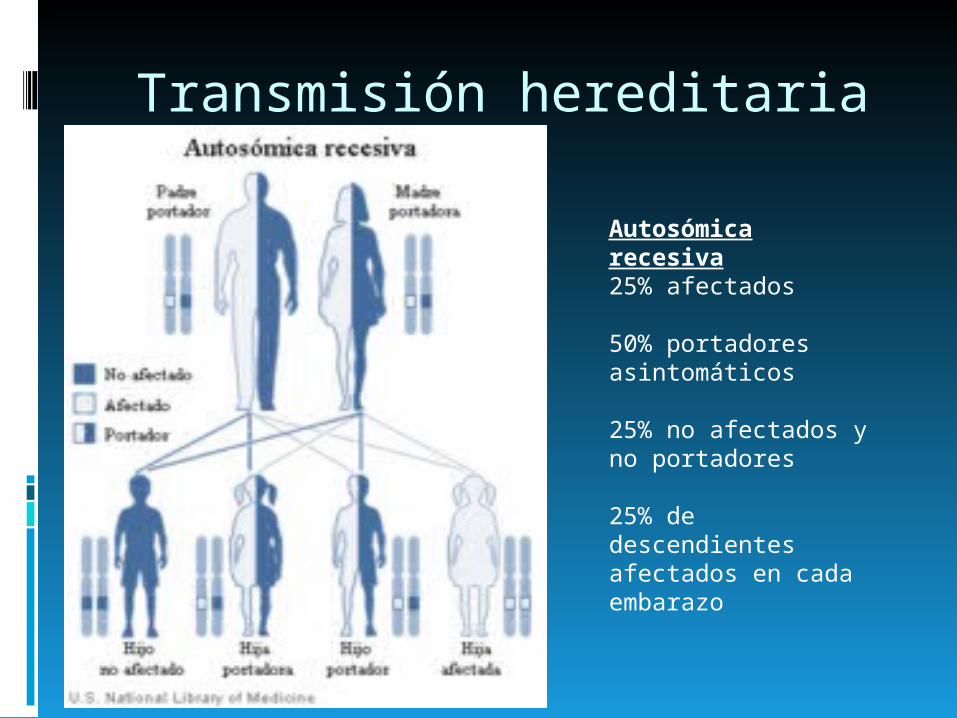
Transmisión hereditaria
Autosómica recesiva25% afectados
50% portadores asintomáticos
25% no afectados y no portadores
25% de descendientes afectados en cada embarazo

Diagnostico
El diagnóstico de talasemias está basado en análisis:
clínico hematológico
molecular

Diagnóstico clínico
Talasemia mayor: en niños menores de 2 años, anemia microcítica severa, ictericia leve y hepatoesplenomegalia
Talasemia intermedia: en niños de edad mas avanzada con cuadros clínicos similares.
Talasemia menor: En general son asintomáticos y pueden presentar una anemia muy leve.
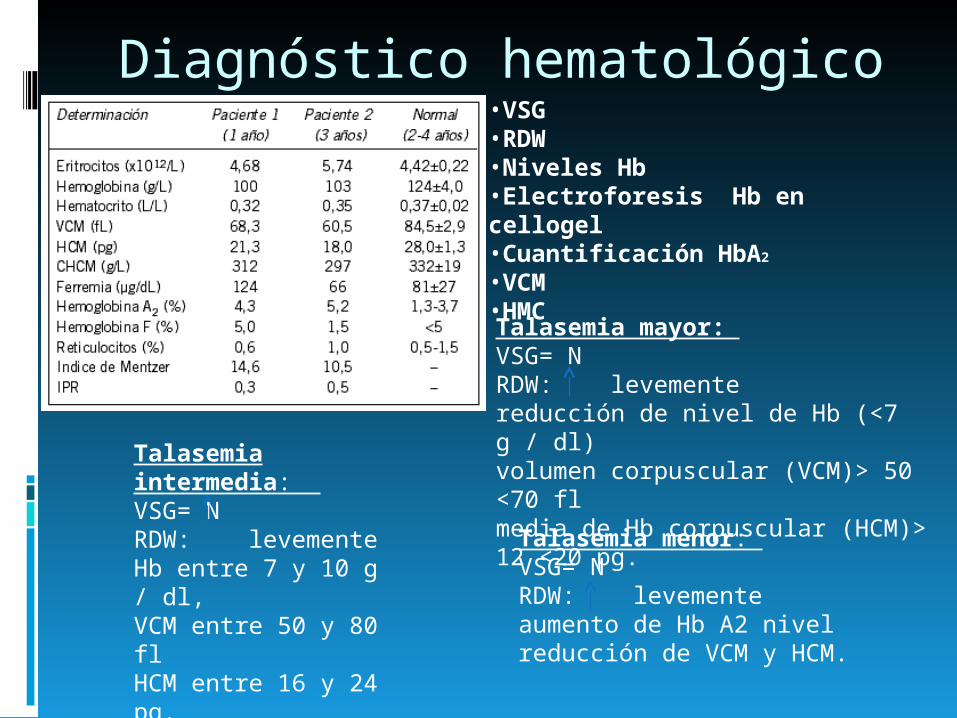
Diagnóstico hematológico
Talasemia intermedia: VSG= NRDW: levementeHb entre 7 y 10 g / dl,VCM entre 50 y 80 flHCM entre 16 y 24 pg.
Talasemia mayor: VSG= NRDW: levementereducción de nivel de Hb (<7 g / dl)volumen corpuscular (VCM)> 50 <70 flmedia de Hb corpuscular (HCM)> 12 <20 pg. Talasemia menor:
VSG= NRDW: levementeaumento de Hb A2 nivelreducción de VCM y HCM.
•VSG•RDW•Niveles Hb•Electroforesis Hb en cellogel•Cuantificación HbA2 •VCM•HMC
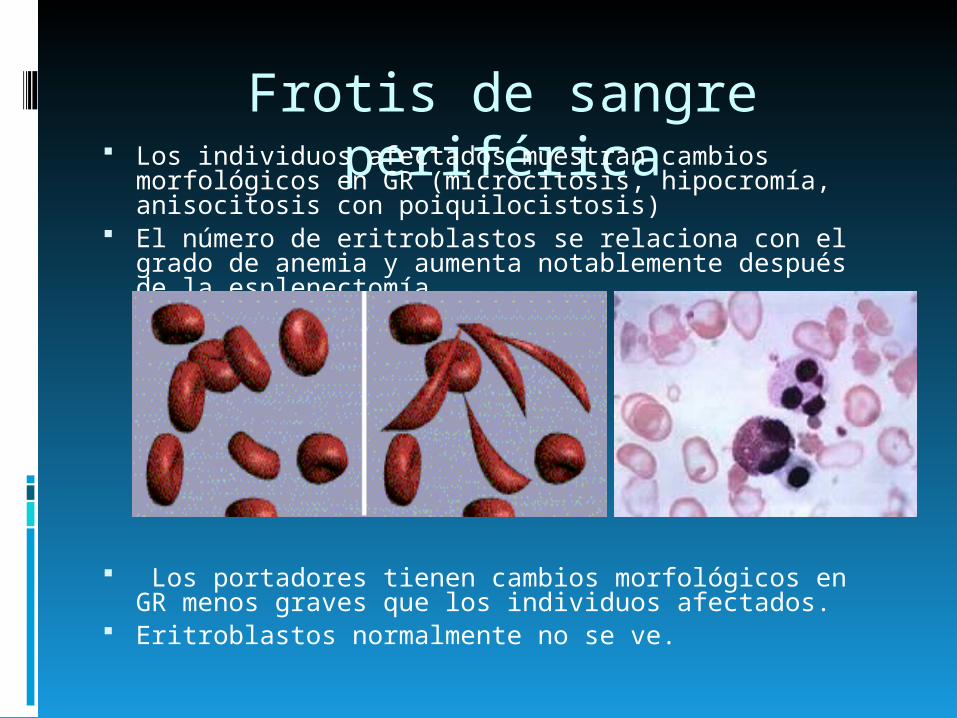
Frotis de sangre periférica Los individuos afectados muestran cambios
morfológicos en GR (microcitosis, hipocromía, anisocitosis con poiquilocistosis)
El número de eritroblastos se relaciona con el grado de anemia y aumenta notablemente después de la esplenectomía.
Los portadores tienen cambios morfológicos en GR menos graves que los individuos afectados.
Eritroblastos normalmente no se ve.

Análisis cualitativo y cuantitativo de Hb
Determinación de la concentración de Hb en sangre total, mediante el método de cianometaHb
Electroforesis en cellogel: indica la cantidad y tipo de Hb presente.
El patrón de la Hb en β-talasemia varía: β0 talasemia homocigotos(mayor): HbA está ausente HbF constituye el 92-95% del total de Hb. β+ homocigotos y β+/β0 heterocigota : HbA están entre el 10 y el 30% HbF entre 70-90%. HbA2 es variable en BT homocigota y se
ha mejorado en la beta talasemia menor.

Electroforesis de Hb sobre cellogel
Valores normales:•Hb A > 97%•Hb A2 < 3%•Valores mayores de Hb A2 indican BT heterocigota
Separación de hemo-proteinas
Cuantificación
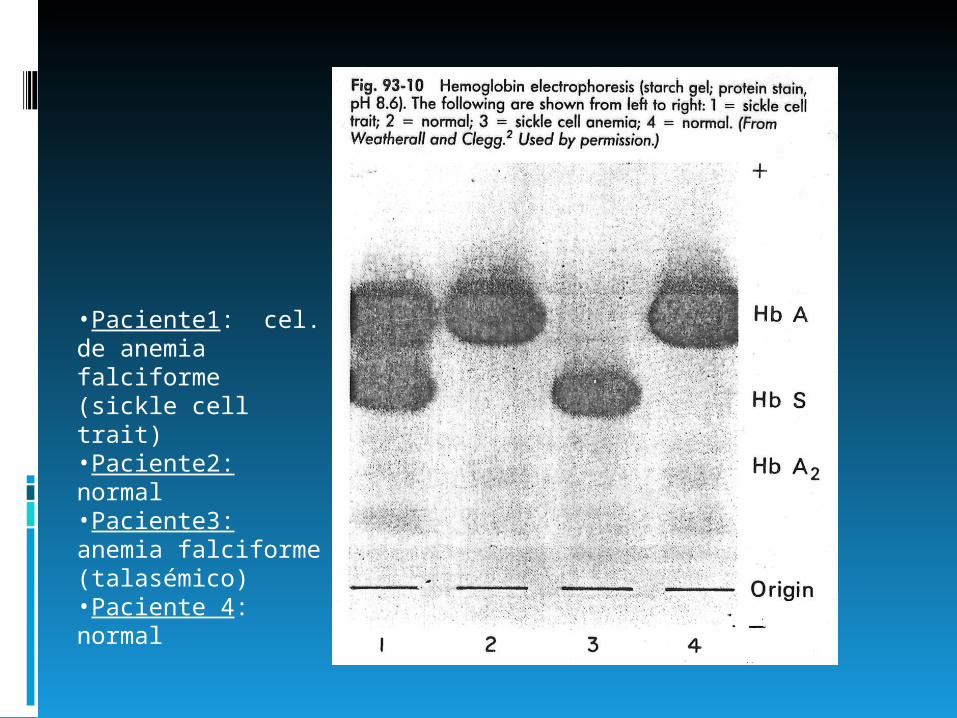
•Paciente1: cel. de anemia falciforme (sickle cell trait)•Paciente2: normal•Paciente3: anemia falciforme (talasémico)•Paciente 4: normal

Diagnóstico molecular
Las mutaciones son detectadas con el siguiente procedimiento:
Extracción de ADN
PCR
Dot-Blot (ASO)
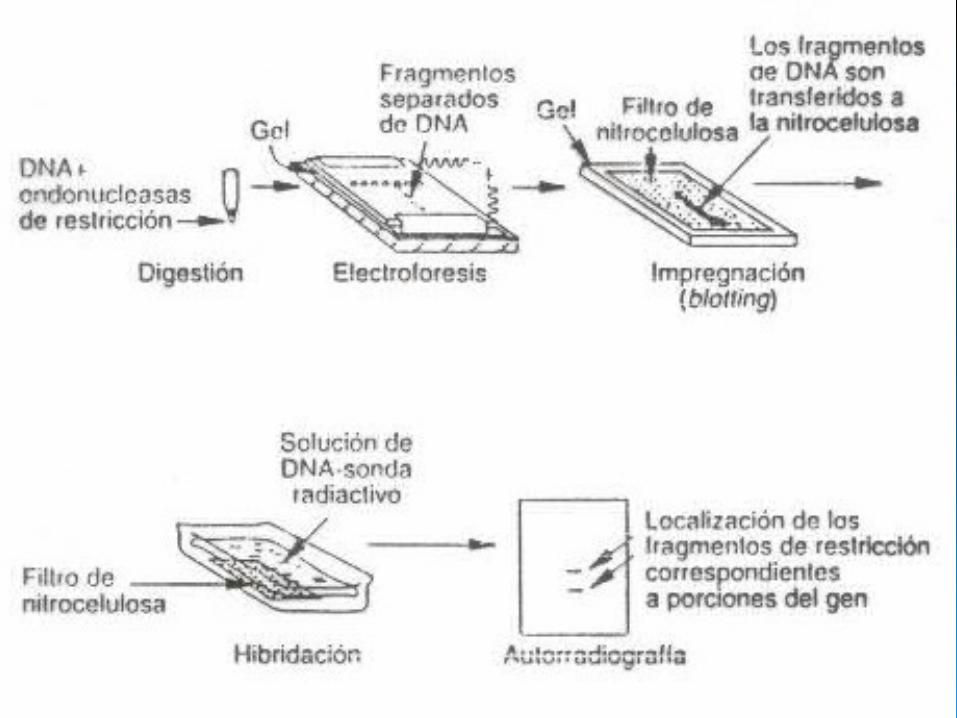

Diagnóstico
El diagnostico diferencial es generalmente sencillo:
anemias sideroblásticas: anillo perinuclear, C de protoporfirinas eritrocíticas
anemias desideropoyeticas congénita: no tiene elevada HbF, multinuclearidad de precursores de GR.
trastornos con altos niveles de HbF Asesoramiento genético y diagnostico
prenatal.

Tratamiento
Trasplante de médula ósea (cura definitiva)
Trasplante de sangre de cordón umbilical
(cura definitiva)

Tratamiento Transfusiones:
valores de Hb caen por debajo de 7gr/dl deformaciones facialescrecimiento pobreexpansión óseaaumento de esplenomegalia.

Tratamiento Transfusiones:
Cantidad de sangre que se transfunde depende: peso del paciente nivel de Hb hematocrito etc.

Tratamiento Transfusiones:
frecuencia de 2 a 4 semanas.
La cantidad transfundida no debe exceder de 15 a 20 ml/kg/día; infundida a una Vmax 5ml/kg/hr

Manifestaciones clínicas de la sobrecarga de Fe
Hipogonadismo Hipotiroidismo Hipoparatiroidismo Diabetes Osteoporosis Disfunción cardíaca (principal causa de muerte) Fibrosis hepática
hormonas sexuales
L-tiroxina
Calcio y vit D
Acarbosa
Prevencion

Manifestaciones clínicas de la sobrecarga de Fe
Control de los niveles de Fe en sangre:
Ferritina sérica Resonancia Magnética
Nuclear Biopsia de hígado (muy
invasiva)

Tratamiento de la sobrecarga de Fe
Esplenectomía
Terapia de quelación: Administra quelantes de Fe
para eliminación por orina y heces (a partir de 10 a 20 transfusiones o a
niveles de ferritina por encima de 1000 ng/ml)

Tratamiento Fármacos disponibles: deferoxamina (DFO) terapia
combinada deferiprona (DFP) deferasirox (DFX)
(s)-3-(OH)-desazadesferrithiocien-poliéter

Marquez, Ma. Soledad Matkovic, Lucas
Luna, Jaime Casas, josé Ignacio
Fernández Fliguer, Alan