1º congreso bioquímico del litoral - fundatal...dra. margarita bragós cátedra hematología...
TRANSCRIPT

Dra. Margarita Bragós
Cátedra Hematología
FCByF. UNR
Santa Fe, 9, 10 y 11 de junio de 2011
1º Congreso Bioquímico del Litoral
DIAGNÓSTICO
MOLECULAR:
APLICACIONES EN
HEMATOLOGÍA

Talasemia (+, º)
Talasemia (+, º)
Hb F aumentada ()º th,
HPFH th
Talasemias
DIAGNÓSTICO MOLECULAR DE TALASEMIAS
IMB

DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
Se estima que el 3% de la población mundial porta un gen para talasemia
IMB

10 % 20 %
10 %
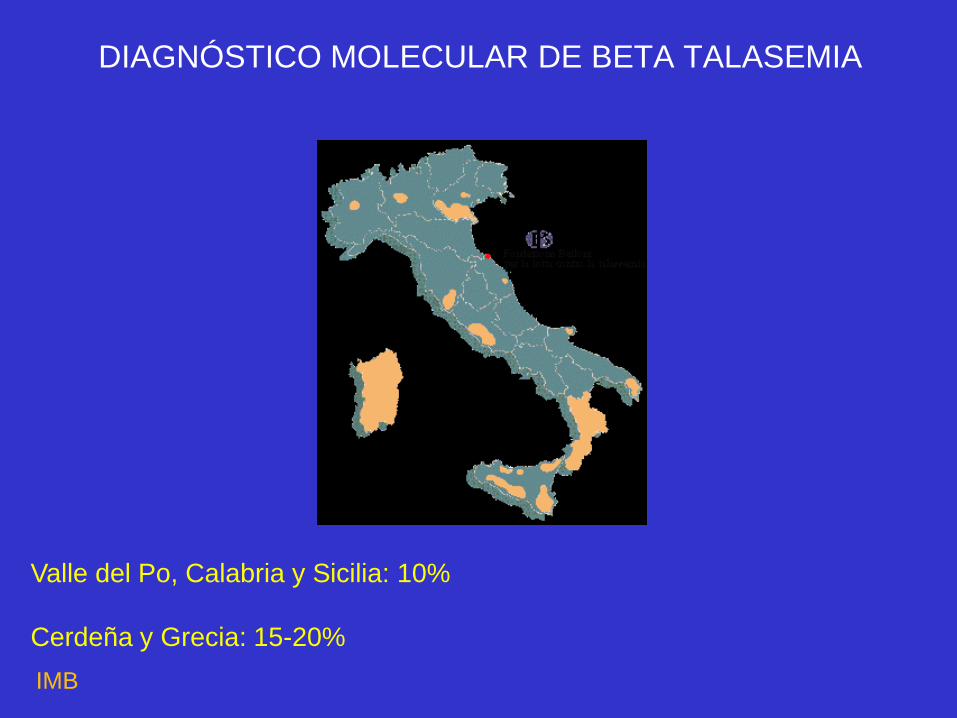
DISTRIBUCIÓN GEOGRÁFICA DE th
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
Valle del Po, Calabria y Sicilia: 10%
Cerdeña y Grecia: 15-20%
IMB

Hb S
10-20 %
< 5 %
40 %
> 20 %
5 % 15 %
DISTRIBUCIÓN GEOGRÁFICA DE HEMOGLOBINOPATÍAS
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

HERENCIA
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
Datos de nuestro país proporcionados por FUNDATAL
Registro a partir de 1997.
Talasemias mayores: 140
Talasemias mayores c/TMO: 17
Talasemias intermedias: 45
HbS/Talasemia y otras: 55
HBSS: 14
IMB
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
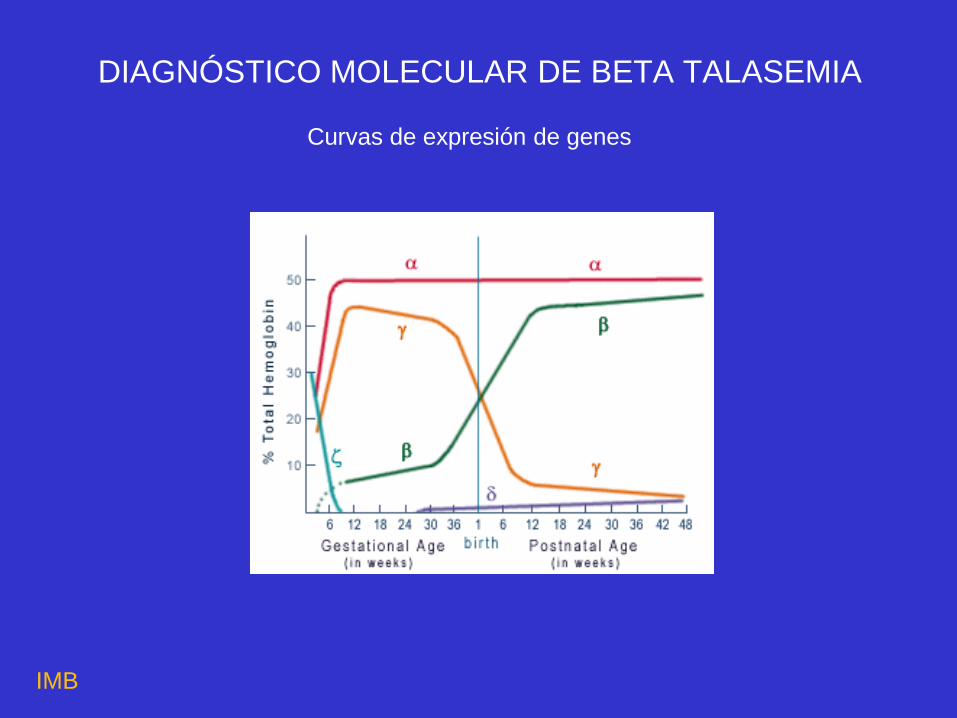
Curvas de expresión de genes
IMB

Clusters de y Globinas
Patrinos Hum Mut, 2005
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

Mutaciones en Talasemia
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
Esquema del proceso de biosíntesis de la cadena globina. En la fase de Transcripción se
sintetiza el pre-ARNm en el interior del núcleo. Durante el Procesamiento se modifica y acorta el
transcripto primario (splicing). En el citoplasma, por el proceso de Traducción, el ARNm maduro
se pega a los ribosomas para comenzar la síntesis proteica.
IMB

Frameshift º º Splicing
Promotor
RNA cleavage
Small deletion
+
+ º º º º º º º º º º
º º
º º º º º º º º º º º º º º
º º º º º º º º * * * * * * * * * *
8 ag 8 ag 7 gt
7 gt
Cap site
* * Unstable Globin
Nonsense mutation
+ Initatiation codon
Mutaciones en Talasemia
1 30 31 104 105 146
5' 3'
LCR LCR
G A 3’ HS1 3’ HS1
Defectos en la transcripción del RNA
Defectos en el procesamiento del RNA
Defectos en la traducción del RNA
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

Mutaciones en Talasemia
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

Mutaciones en Talasemia
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

Mutaciones en Talasemia
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
MUTACIONES QUE AFECTAN AL PROCESAMIENTO DEL ARNm
A.- Mutación en el sitio de unión del CAP: una sola mutación ha sido
encontrada en esta posición.
B.- Mutaciones en el proceso de empalme (splicing)
1. - Mutaciones en las secuencias donadora o aceptora para el splicing:
IVS1-1 GA, IVS2-l GA (0 –talasemia).
2. - Mutaciones en los exones (mutación sin sentido): Hb Malay (19 GA),
HbE (26 G A) y Hb Knossos (27 G T) (+ -talasemia)
3. - Mutaciones en los intrones que originan un lugar alternativo para el
splicing: IVS1-6 TC, IVS1-110 GA; IVS2-745 (+ talasemia). ,
C.- Mutaciones que afectan a la escisión y poliadenilación
IMB

Mutaciones en Talasemia
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
MUTACIONES EN EL PROCESO DE TRADUCCIÓN
A.- Codon de iniciación: en la posición 49 a la izquierda del sitio CAP,
aparece un codon AUG que corresponde a la señal para iniciar la
traducción. Las posibles mutaciones que se originen en este codon,
suprimen la traducción y originan una 0-talasemia.
B.- Terminación prematura
1. - Mutación sin sentido: la sustitución de una de las bases de un triplete
que codifica a un aminoácido, origina uno de los tres codones de
terminación (UAG, UAA y UGA) lo cual da lugar a la terminación
prematura de la traducción del ARNm. CAGTAG en el codon 39 (0 –
talasemia).
2. - Frameshift (Alteración del marco de lectura): inserciones o deleciones
de 1, 2 o 4 nucleótidos en la región codificadora del gen globina (0 –
talasemia). IMB

DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
CORRELACIONES GENOTIPO-FENOTIPO
Cualquier condición heredada o adquirida que reduce el desbalance de
cadenas de globina alpha/no-alpha en ß-talasemia resulta en un menor
grado de precipitación de cadenas de globina alfa y conduce a un fenotipo
menos manifiesto [Galanello & Cao 1998].
•El cuadro clínico que resulta de la homocigosidad ß+ -talasemia u
homocigosidad para ߺ-talasemia puede ser mejorado por la co-herencia
de mutaciones en el gen que codifica la cadena de alfa globina, lo que
reduce la producción cadenas de alfa globina y por lo tanto disminuye la
relación alpha/no-alpha.
•ß-talasemia Heterocigota puede en algunas circunstancias dar una
fenotipo de talasemia intermedia en lugar del carrier asintomático.
Triplicación o (menos frecuentemente) cuadruplicación del gen alpha
(/ o / o /), lo que aumenta el desbalance en la
relación de cadenas alpha/no-alpha.
IMB
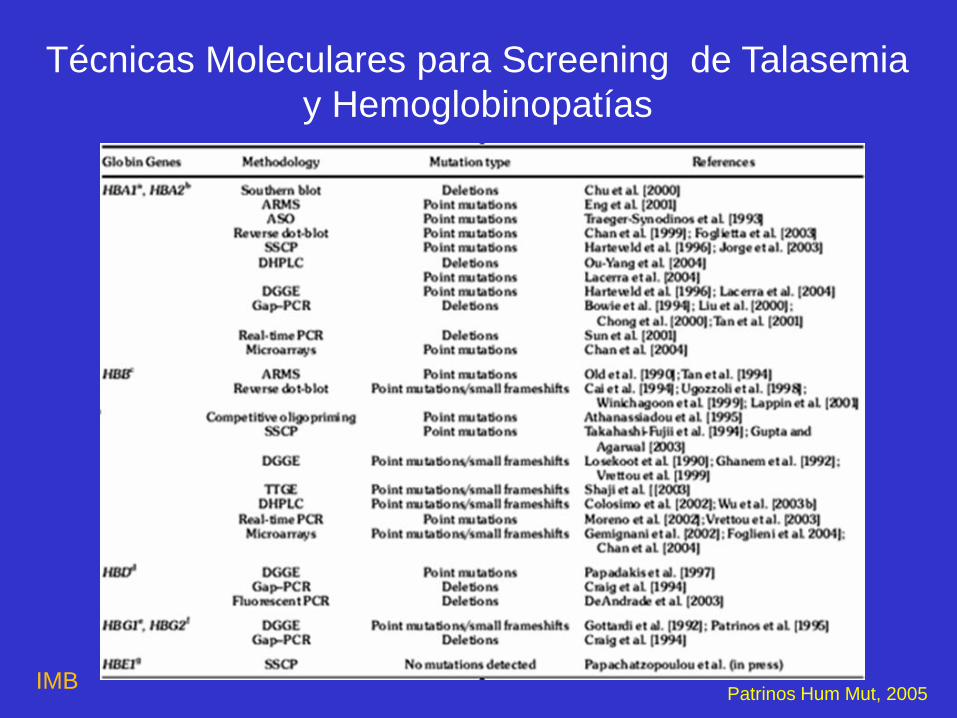
Técnicas Moleculares para Screening de Talasemia
y Hemoglobinopatías
Patrinos Hum Mut, 2005 IMB
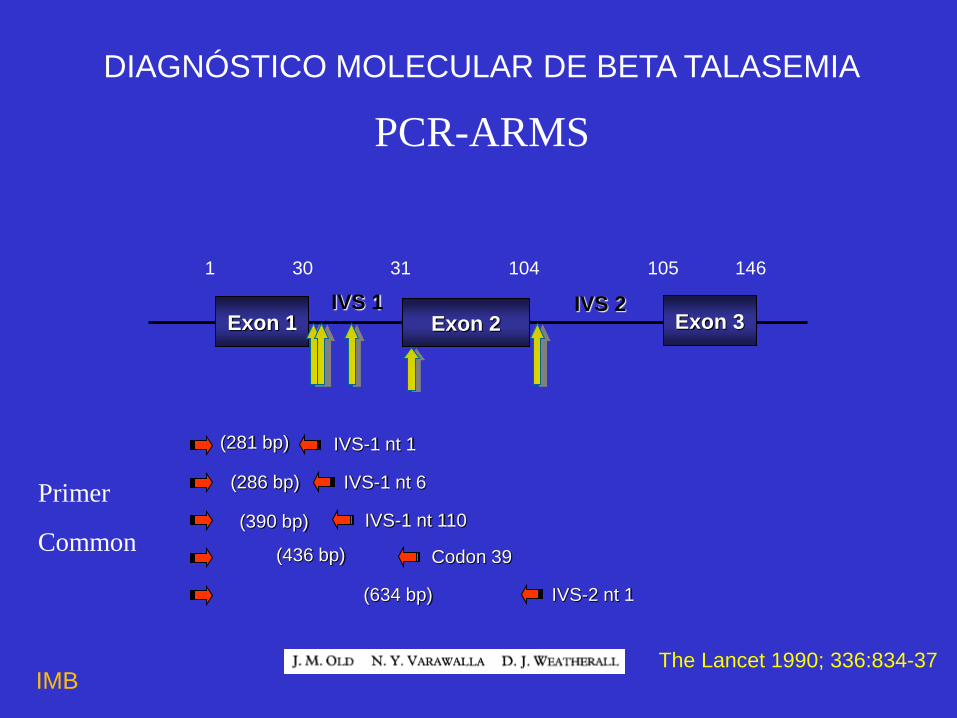
Exon 1 Exon 2 Exon 3 IVS 1 IVS 2
1 30 31 104 105 146
(634 bp) IVS-2 nt 1
(436 bp) Codon 39
(390 bp) IVS-1 nt 110
(286 bp) IVS-1 nt 6
(281 bp) IVS-1 nt 1
Primer
Common
PCR-ARMS
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
The Lancet 1990; 336:834-37 IMB

+ 1-110 / 0 39 Talasemia
1 2 3 4 1 2 3 4
Línea 1: control de contaminación, H2O
Líneas 2 y 4: 100 bp ladder
a- Líneas 3 y 4: doble-heterocigota IVS-1-nt110 con primer normal y mutado (390 bp)
b- Líneas 2 y 3: doble-heterocigota CD39 con primer normal y mutado (436 pb).
a b
500bp
861bp
390bp
861bp
436bp
IVS-1-nt110 CD39
PCR-ARMS
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

- Talasemia
DOT-BLOT
B2, C1 y D1 son heterocigotas para IVS 1-110
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
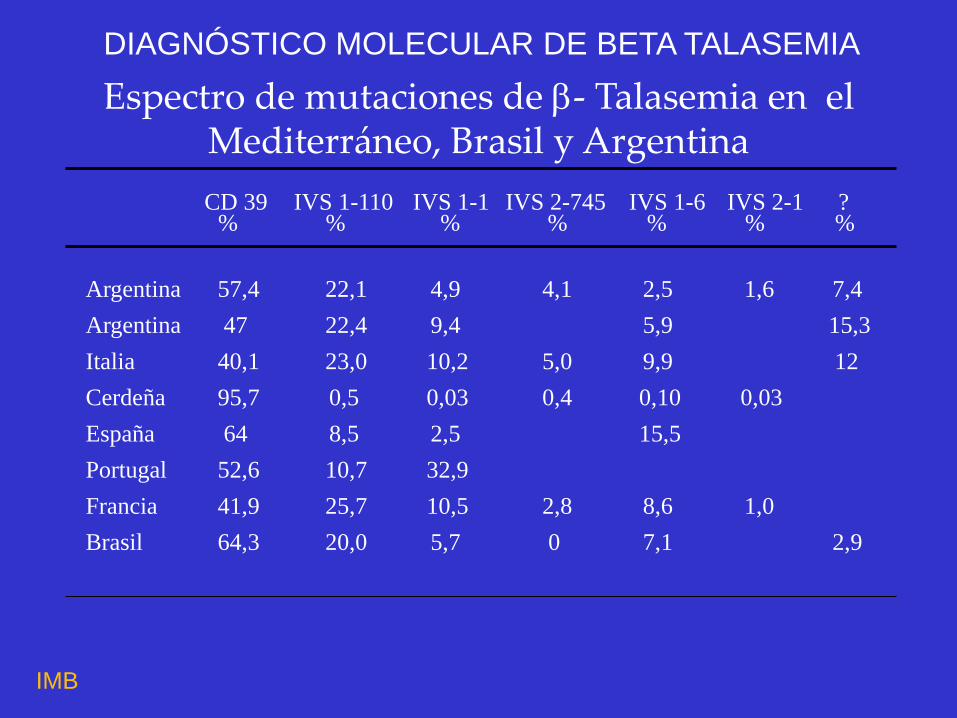
Espectro de mutaciones de - Talasemia en el Mediterráneo, Brasil y Argentina
CD 39 %
IVS 1-110 %
IVS 1-1 %
IVS 2-745 %
IVS 1-6 %
IVS 2-1 %
? %
Argentina 57,4 22,1 4,9 4,1 2,5 1,6 7,4
Argentina 47 22,4 9,4 5,9 15,3
Italia 40,1 23,0 10,2 5,0 9,9 12
Cerdeña 95,7 0,5 0,03 0,4 0,10 0,03
España 64 8,5 2,5 15,5
Portugal 52,6 10,7 32,9
Francia 41,9 25,7 10,5 2,8 8,6 1,0
Brasil 64,3 20,0 5,7 0 7,1 2,9
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

Espectro de mutaciones de - Talasemia en Rosario
MUTACIONES Nº DE ALELOS % FENOTIPO
IVS-1-nt l (G A)4,9 6 º
IVS-1-nt 6 (T C)2,5 3 +
IVS-2-nt l (G A)1,6 2 º
IVS-1-nt 110 (G A)22,1 27 +
IVS-2-nt 745 (C G)4,1 5 +
CD39 (C T)57,4 70 º
Desconocidos 9 7,4
Total 122 100
Origen de los pacientes: 89,3 % italianos; 9,8 % españoles; 0,9 % griegos
Bragós et al. Haematologica, 2000
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

HETEROGENEIDAD MOLECULAR DE LA -TALASEMIA
Autores Mutaciones
estudiadas
% de alelos
identificados
Bragós, I 6 92,6
Rosatelli, MC (Italia) 5 88
Martins, C (Brasil) 4 97,1
Villegas, A (España) 5 86,6
Varela, V
(Argentina)´96
4 90
Varela, V
(Argentina)´99
6 90
Roldán, A
(Argentina)
4 84,7
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
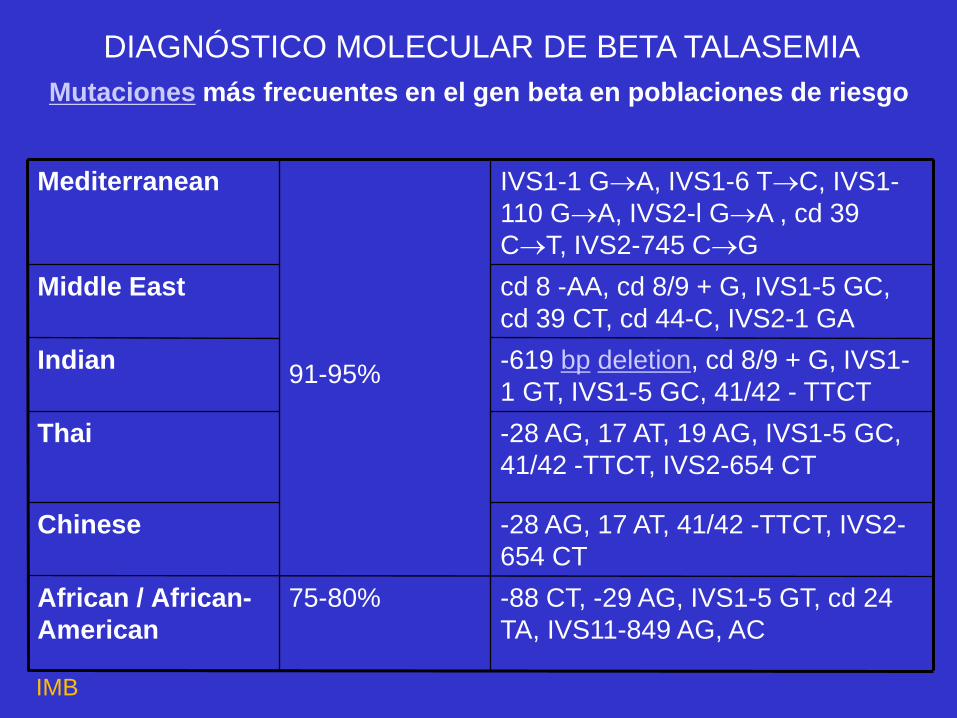
Mutaciones más frecuentes en el gen beta en poblaciones de riesgo
-88 CT, -29 AG, IVS1-5 GT, cd 24
TA, IVS11-849 AG, AC
75-80%
African / African-
American
-28 AG, 17 AT, 41/42 -TTCT, IVS2-
654 CT
Chinese
-28 AG, 17 AT, 19 AG, IVS1-5 GC,
41/42 -TTCT, IVS2-654 CT
Thai
-619 bp deletion, cd 8/9 + G, IVS1-
1 GT, IVS1-5 GC, 41/42 - TTCT
91-95% Indian
cd 8 -AA, cd 8/9 + G, IVS1-5 GC,
cd 39 CT, cd 44-C, IVS2-1 GA
Middle East
IVS1-1 GA, IVS1-6 TC, IVS1-
110 GA, IVS2-l GA , cd 39
CT, IVS2-745 CG
Mediterranean
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
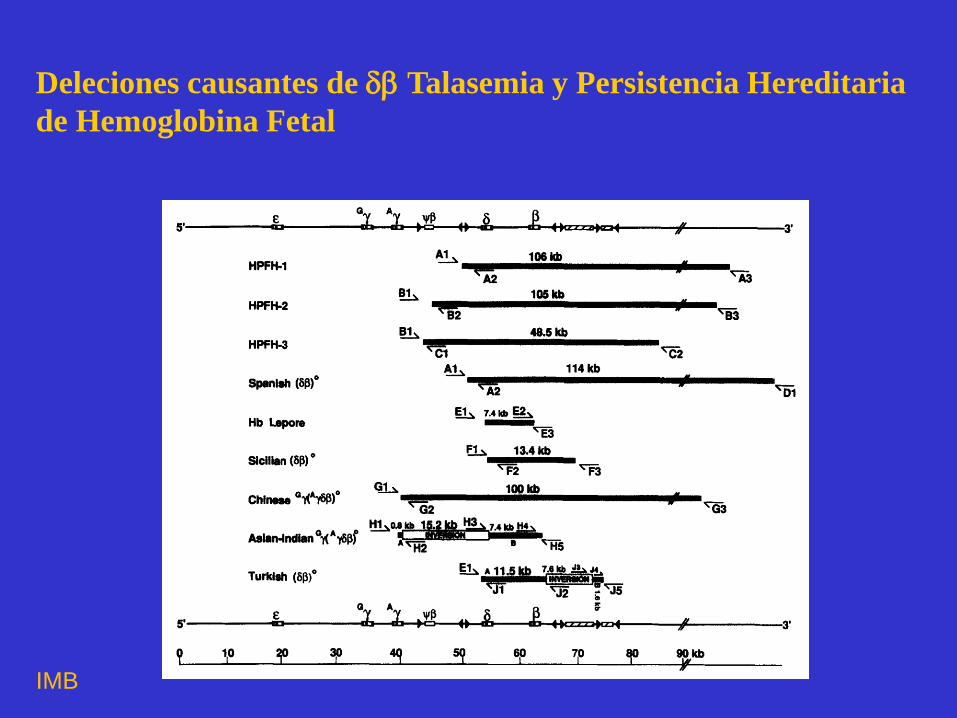
Deleciones causantes de Talasemia y Persistencia Hereditaria
de Hemoglobina Fetal
IMB

()0 Siciliana
1- Control normal
2- Control positivo heterocigota
3- Paciente positivo heterocigota
4- Blanco de reacción
5- Marker
1 2 3 4 5
1585 pb
1150 pb
IMB

Talasemia Intermedia. Co-herencia de triplicación de genes α y th
(a) + II-745/ anti 3,7 (b) 0 39/ anti 3,7
32.2 17.1 53 27 8.6 (b) 5.0
30.9 15.8 51 28.5 8.8 (a) 5.7
MCHC
(g/dl)
MCH
(pg)
MCV
(fl)
Hct
(%)
Hb
(g/dl)
RBC
(1012/l)
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
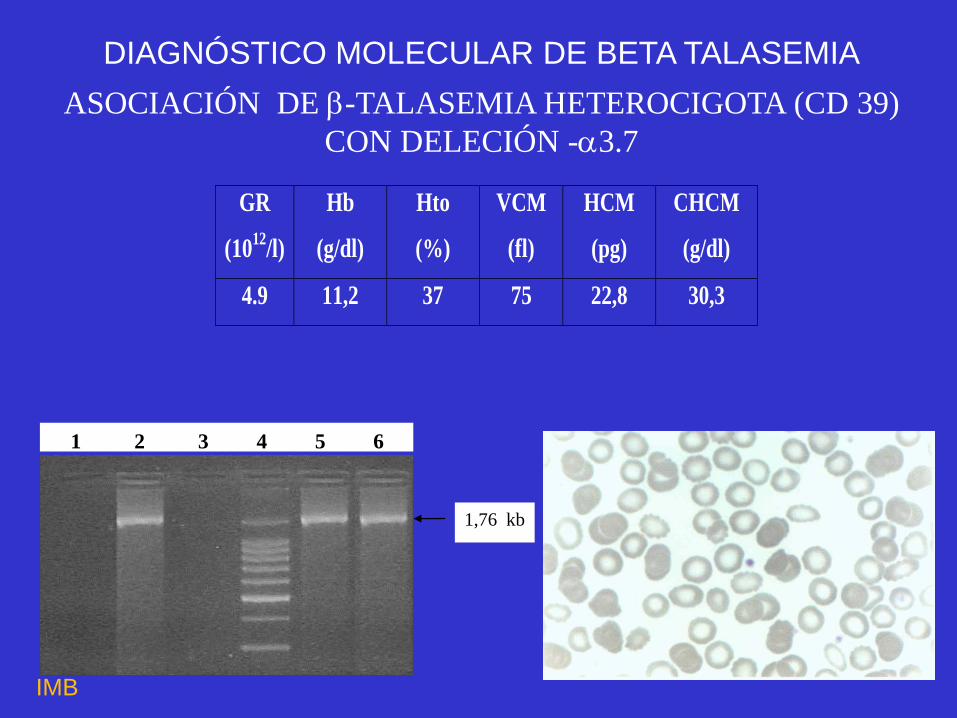
ASOCIACIÓN DE -TALASEMIA HETEROCIGOTA (CD 39)
CON DELECIÓN -3.7
GR
(1012
/l)
Hb
(g/dl)
Hto
(%)
VCM
(fl)
HCM
(pg)
CHCM
(g/dl)
4.9 11,2 37 75 22,8 30,3
1 2 3 4 5 6
1,76 kb
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
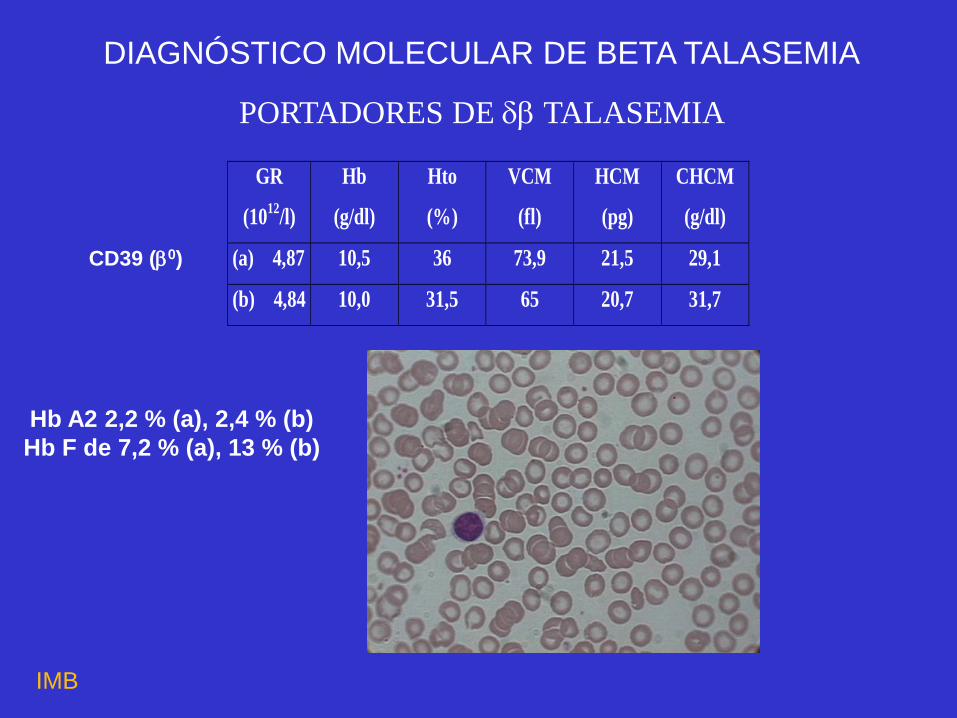
PORTADORES DE TALASEMIA
GR
(1012
/l)
Hb
(g/dl)
Hto
(%)
VCM
(fl)
HCM
(pg)
CHCM
(g/dl)
(a) 4,87 10,5 36 73,9 21,5 29,1
(b) 4,84 10,0 31,5 65 20,7 31,7
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
CD39 (0)
Hb A2 2,2 % (a), 2,4 % (b)
Hb F de 7,2 % (a), 13 % (b)
IMB
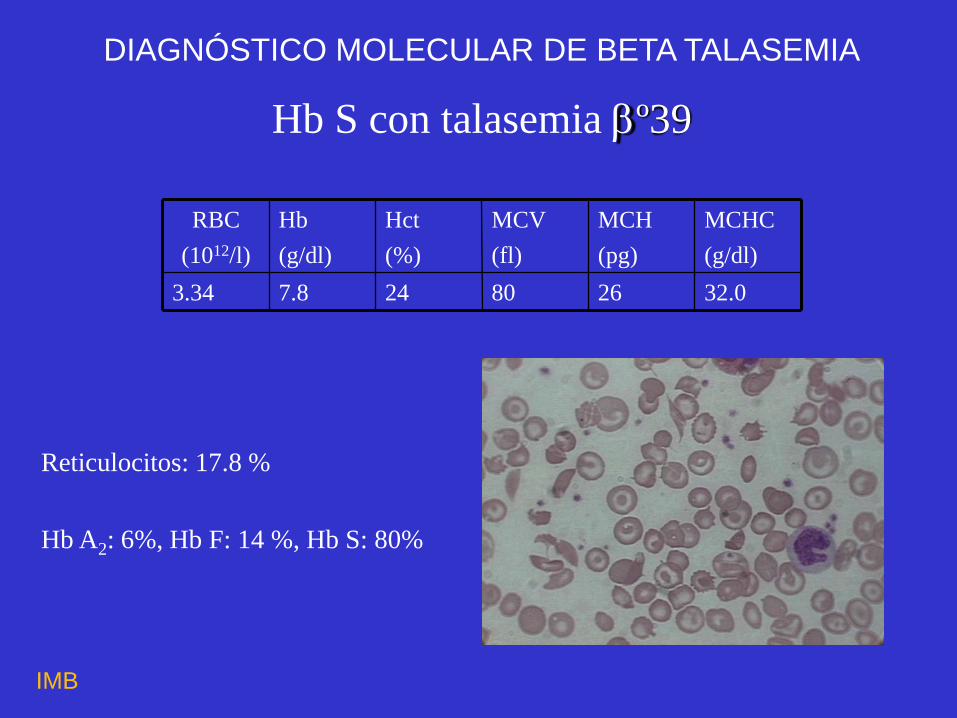
Hb S con talasemia º39
32.0 26 80 24 7.8 3.34
MCHC
(g/dl)
MCH
(pg)
MCV
(fl)
Hct
(%)
Hb
(g/dl)
RBC
(1012/l)
Reticulocitos: 17.8 %
Hb A2: 6%, Hb F: 14 %, Hb S: 80%
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB
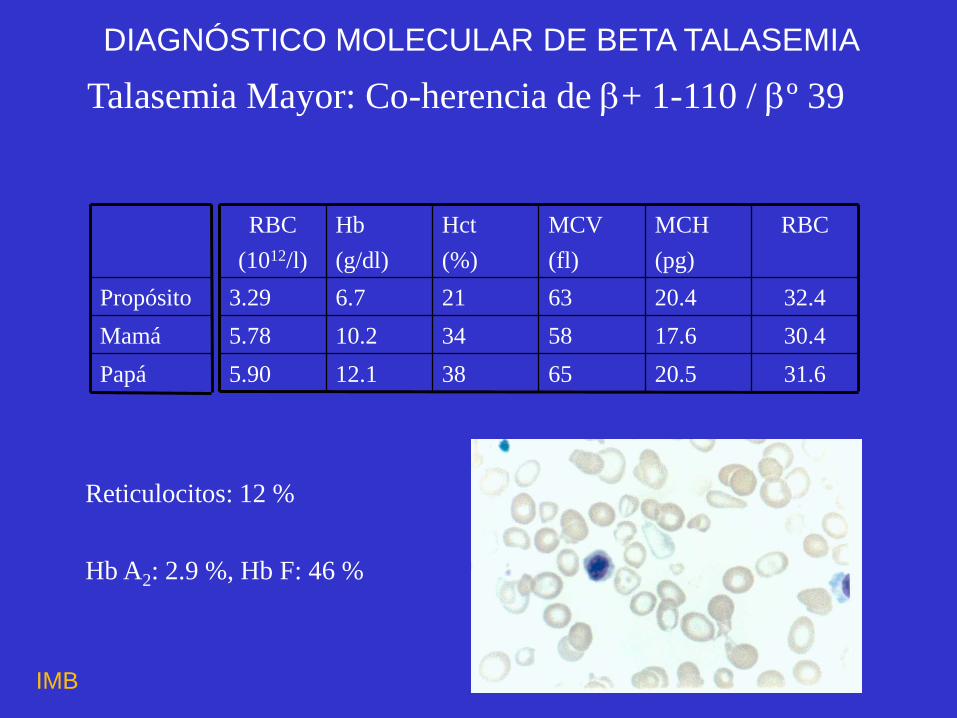
Talasemia Mayor: Co-herencia de + 1-110 / º 39
Reticulocitos: 12 %
Hb A2: 2.9 %, Hb F: 46 %
31.6 20.5 65 38 12.1 5.90
30.4 17.6 58 34 10.2 5.78
32.4 20.4 63 21 6.7 3.29
RBC MCH
(pg)
MCV
(fl)
Hct
(%)
Hb
(g/dl)
RBC
(1012/l)
Papá
Mamá
Propósito
DIAGNÓSTICO MOLECULAR DE BETA TALASEMIA
IMB

Clusters de y Globinas
Patrinos Hum Mut, 2005
DIAGNÓSTICO MOLECULAR DE ALFA TALASEMIA
IMB

80 %
5 - 10 %
20 - 30 %
80 %
DISTRIBUCIÓN GEOGRÁFICA DE th
DIAGNÓSTICO MOLECULAR DE ALFA TALASEMIA
IMB

α TALASEMIA
Los alelos talasémicos (-/) (--/) (T/) de los progenitores
portadores se combinan en sus diferentes posibilidades dando
distintos genotipos
IMB

α TALASEMIA
Los alelos talasémicos (-/) (--/) (T/) de los progenitores
portadores se combinan en sus diferentes posibilidades dando
distintos genotipos
IMB

Diagnóstico Molecular de Talasemias
DIAGNÓSTICO MOLECULAR DE ALFA TALASEMIA
IMB
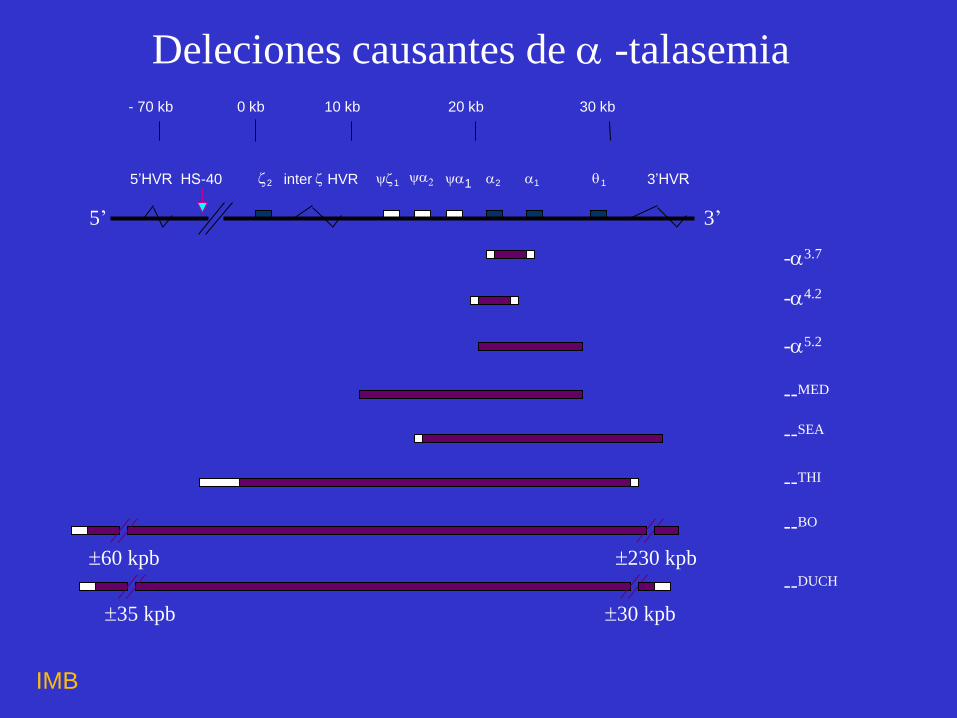
-4.2
-3.7
- 70 kb 0 kb 10 kb 20 kb 30 kb
5’HVRHS-40 2 inter HVR 1 1 2 1 1 3’HVR
5’ 3’
-5.2
--MED
--THI
--SEA
--BO
230 kpb 60 kpb
30 kpb 35 kpb
--DUCH
Deleciones causantes de -talasemia
IMB

- 70 kp 0 kp 10 kp 20 kp 30 kp
5’HVRHS-40 2 inter HVR 1 1 2 2 1 1 3’HVR
Nco Hph CS T Saudí
CAP Poly A signal
1 31 32 99 100 141
5' 3'
CAT ATA box box
1) Mutaciones en el procesamiento del RNA
Mutaciones puntuales causantes de -Talasemia
2) Mutaciones en la traducción del RNA
3) Mutaciones que causan
inestabilidad post-transcripcional
Mutaciones en el sitio de splicing
Mutaciones en la señal de poliadenilación
Mutaciones en el codon de iniciación
Mutaciones en el codon de terminación
Mutaciones Frameshift
Mutaciones sin sentido
IMB

Hph I Hph I Hph I Hph I
2
1
C8 C3
ARNm Cap site
1078
163 322
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB

Hph I Hph I Hph I
2
1
C8 C3
ARNm Cap site
1400
163
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB

Nco I
2
1
C8 C3
ARNm Cap site
1094 894
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB

2
1
C8 C3
ARNm Cap site
1998
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB

2
1
C8 C3
ARNm Cap site
Mse I Mse I Mse I
1379 401
163
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB

2
1
C8 C3
ARNm Cap site
Mse I Mse I Mse I
1379 401
163
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB
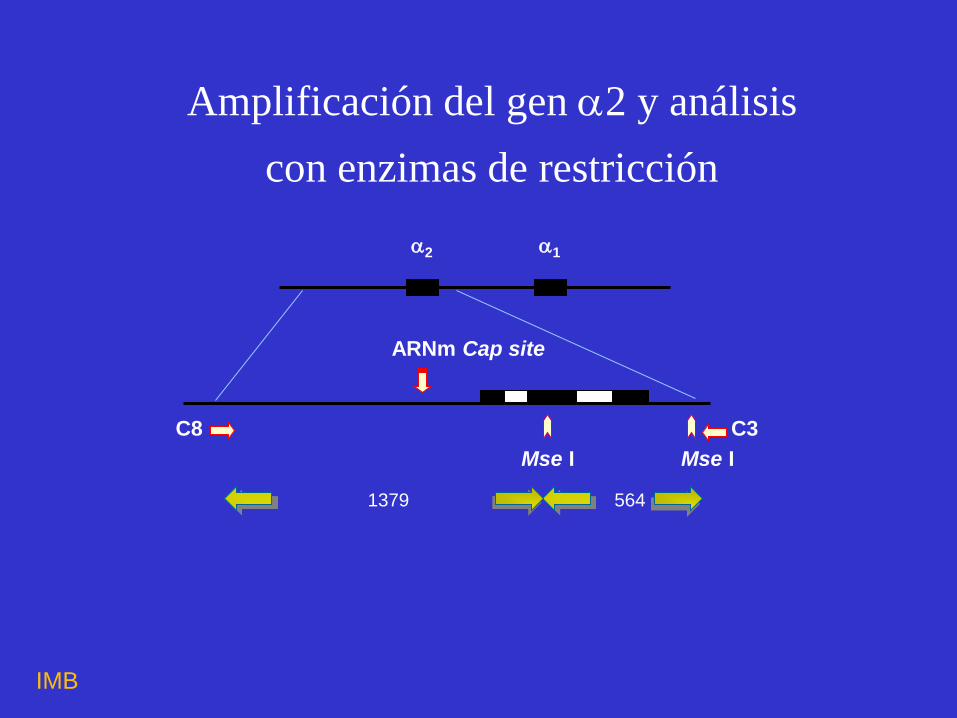
564
2
1
C8 C3
ARNm Cap site
Mse I Mse I
1379
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB

2
1
C8 C3
ARNm Cap site
Mse I Mse I Mse I
1379 401
163
Amplificación del gen 2 y análisis
con enzimas de restricción
IMB
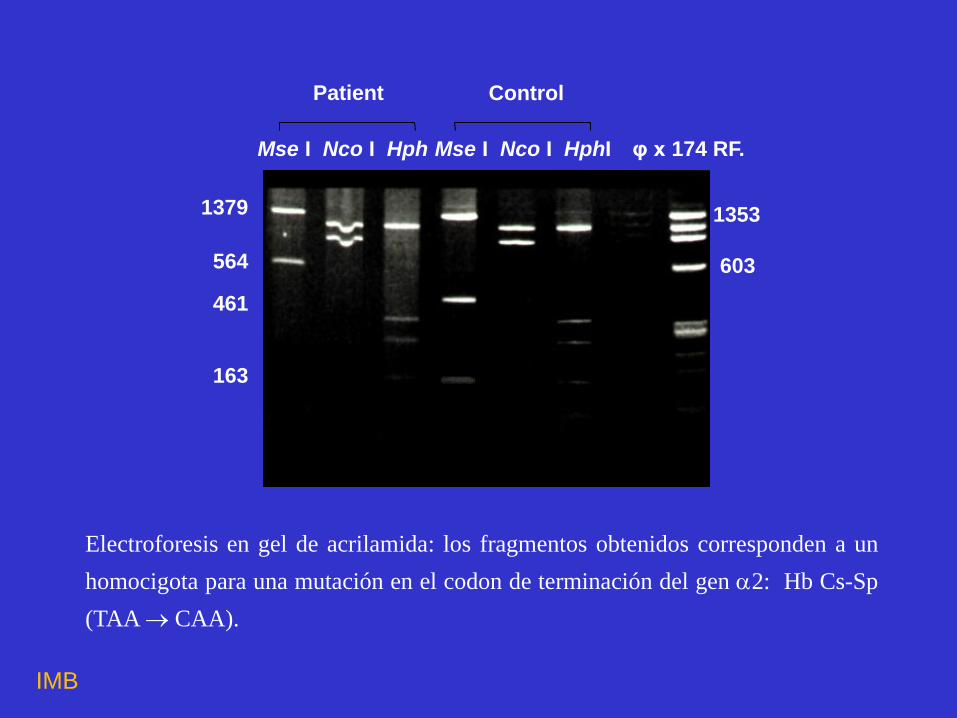
1379
564 461 163
Mse I Nco I Hph Mse I Nco I HphI
Patient Control
φ x 174 RF.
1353
603
Electroforesis en gel de acrilamida: los fragmentos obtenidos corresponden a un
homocigota para una mutación en el codon de terminación del gen 2: Hb Cs-Sp
(TAA CAA).
IMB
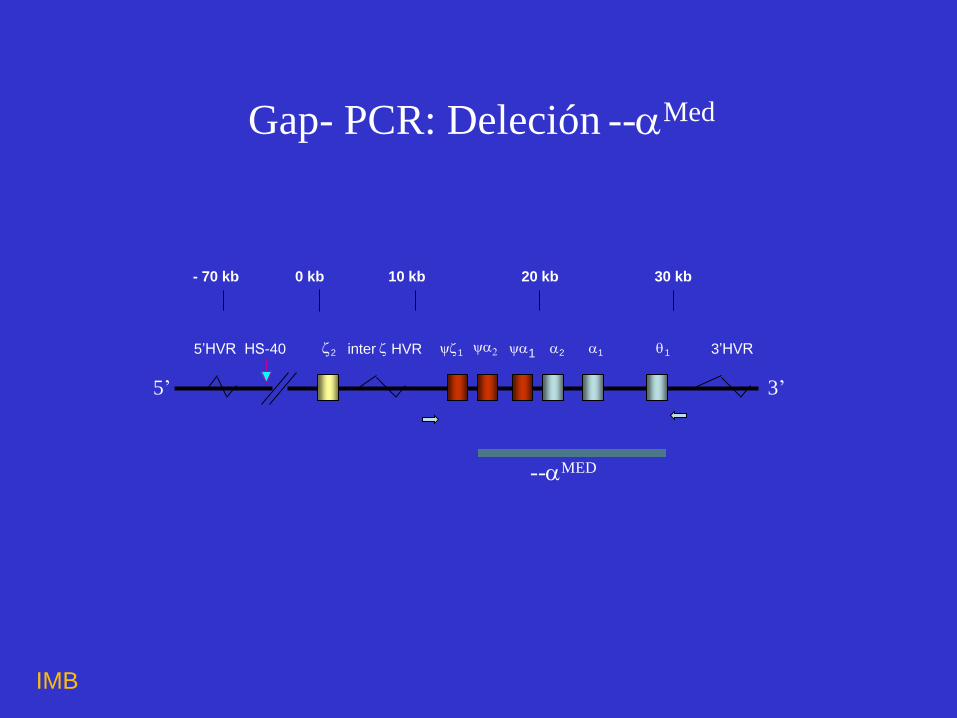
--MED
- 70 kb 0 kb 10 kb 20 kb 30 kb
5’HVRHS-40 2 inter HVR 1 1 2 1 1 3’HVR
5’ 3’
Gap- PCR: Deleción --Med
IMB

1- pUC Mix Marker 8
2– Control Positivo
3- Propósito
4- Mamá
5- Papá
6- Hermano
7- Hermano
861 pb
561 pb
1 2 3 4 5 6 7
1118 pb
692 pb
Deleción --Med
IMB
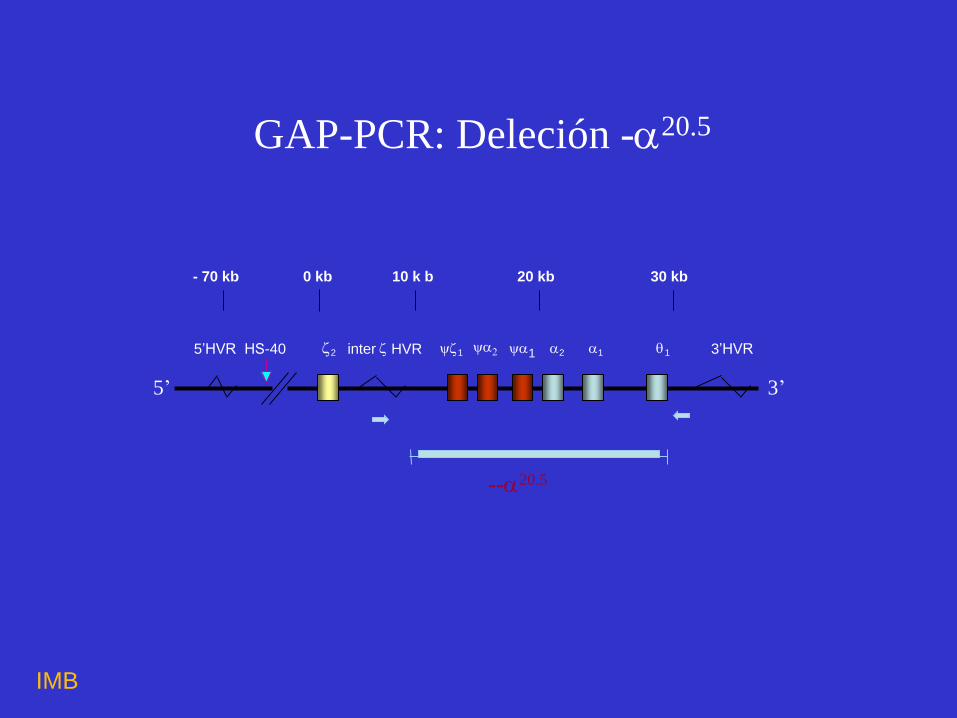
--20.5
- 70 kb 0 kb 10 k b 20 kb 30 kb
5’HVRHS-40 2 inter HVR 1 1 2 1 1 3’HVR
5’ 3’
GAP-PCR: Deleción -20.5
IMB

Deleción -20.5
861 pb 615 pb
1 2 3 4 5 6 7 8
1118 pb
692 pb
1- pGEM DNAMarkers
2- Control Positivo
3- Propósito
4- Mamá
5- Papá
6- Hermano 1997
7- Hermano 1994
8- Control de contaminación
IMB

1 2
5’ 3’
3.7 kb
GAP-PCR: Deleción -3.7
IMB

1 2
5’ 3’
3.7 kb
GAP-PCR: Deleción -3.7
IMB

Deleción -3.7
1-9- pGEM DNAMarker
2-3- Control Positivo
4-5- Propósito
6-7- Mamá
10-11 Papá
12-13 Hermano 1997
14-15 Hermano 1994
8-16 control de contaminación
1.76 kb
1.76 kb
9 10 11 12 13 14 15 16
N M N M N M
N M N M N M
1 2 3 4 5 6 7 8
2.64 kb 1.60 kb
IMB

Morfología eritrocitaria
Mamá Papá
Propósito
IMB

Datos Hematológicos
Aniso++, Micro++,
Hipo++, Elip+,
Ovalo, TC+
141.4
0.6
30.5 / 16
65.8 / 20.0
13.3
6.62
/--20.5
Papá
Aniso+,
Hipo+
4.1
2.0
34.5 / 14.2
81.1/ 28.0
10.8
3.87
/-3.7
Mamá
Aniso+++,
Micro++, Macro+,
Hipo++, Elip+,
Oval+, Esquis+,
TC++
72.4
1.0
31.3 / 22.4
57.7 / 18.0
8.8
4.88
--/-3.7
Propósito
Aniso+
32.6
0.8
33.9 / 12.8
86.6 / 29.3
13.7
4.67
/
Hermano
1997
Aniso+,
Hipo+,
Micro+,
Oval+
44.2
1.9
35.3 / 12.9
79.2 / 28.0
12.8
4.58
/-3.7
Hermano
1994
Genotipo
RBC x 1012/L
Hb g/dl
Reticulocitos %
Morfología
Ferritina
CHCM g/dl /
RW %
MCV fl/ MCH pg
+++: marcada; +: ocasional; -: negativo
hip: hipocromía, aniso: anisocitosis, macro: macrocitos, micro: microcitos, oval: ovalocitos, elip:
eliptocitos, esquis: esquistocitos, TC: target cells
IMB

Prevalencia de talasemia -3.7 en Argentina
Genotipo n Prevalencia
(/) 303 97,74 %
(-3.7/) 6 1,94 %
(-3.7/-3.7) 1 0,32 %
Noguera et al Hemoglobin, 2002 IMB
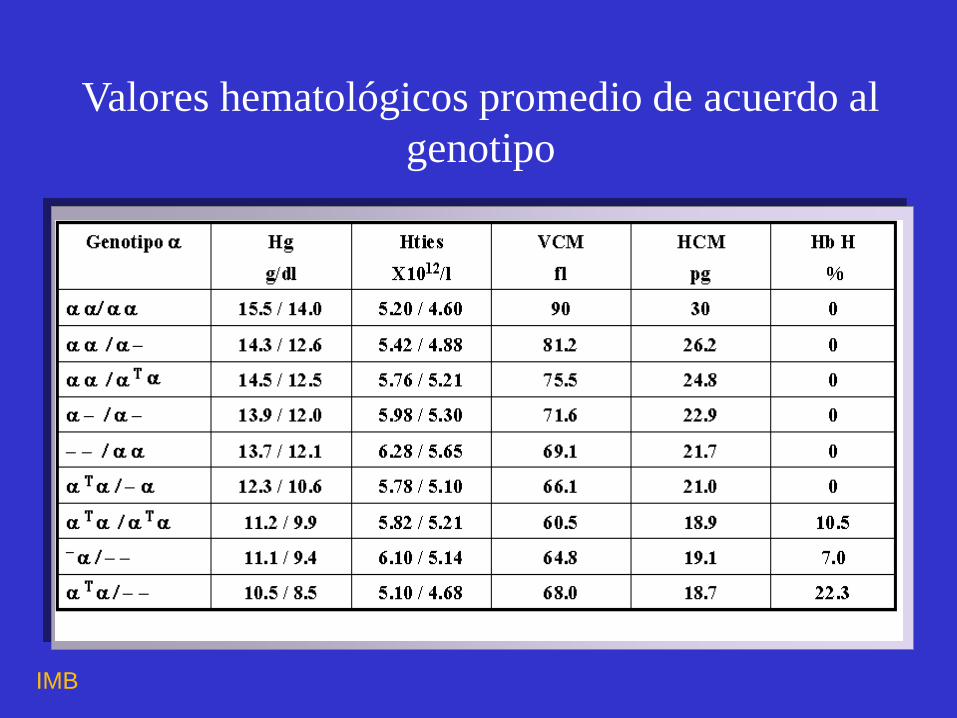
Valores hematológicos promedio de acuerdo al
genotipo
IMB

Desarrollo de una tira ELISA para la detección de talasemias Detección de Hb de Bart en soluciones de Hb mediante mAb altamente
específico .
Sensibilidad (93.4%), especifidad (93,7).
Published online 22 October 2009
Haematologica | 2010; 95(2)
167 sujetos con alfa talasemia
8 muestras positivas por Elisa no pudieron ser
genotipificadas por PCR
IMB

Pruebas de Genética Molecular: Usos Clínicos
•Diagnóstico Molecular para identificar:
oMutaciones puntuales específicas causantes de la enfermedad en el
gen que codifica la cadena beta de la hemoglobina
oDeleciones de extensión variable del gen ß o del cluster, que resultan
en ß-talasemia o en ß-talasemias complejas denominadas ß-talasemia
y ß-talasemia. Nota: las Deleciones son raras causas de ß-talasemia.
•Testeo de Portadores
•Diagnóstico Prenatal
•Pronóstico: predicción de la severidad clínica
DIAGNÓSTICO MOLECULAR DE TALASEMIAS
IMB

El diagnóstico prenatal para identificar la anemia del
Meditarráneo en el feto
-DNA de vellosidad corial: 10-12 semanas de embarazo
-Tener identificadas las mutaciones que afectan a ambos
integrantes de la pareja
-Este estudio permite conocer la situación genética del
feto, pero, en este momento, no hay posibilidad de una
intervención terapéutica para una posible afección fetal.
DIAGNÓSTICO MOLECULAR TALASEMIAS
IMB

Diagnóstico Pre-implantación
Técnica pre-concepción
Recolección de oocitos no fertilizados de la mujer portadora de β-thal. Durante las diferentes fases de maduración, el oocito expele el 1° y el 2°
cuerpo polar. El analisis del DNA de uno de esos cuerpos polares,
implicando que si la mutación talasémica está presente en el cuerpo polar,
no está mas presente en el oocito, permite la selección del oocito sin la
mutación talasémica y por lo tanto la fertilización in vitro para la
implantación en el útero.
DIAGNÓSTICO PRENATAL DE BETA TALASEMIA
IMB

Recientemente se han identificado mutaciones puntuales
responsables de β-talasemia y anemia drepanocítica en
células fetales obtenidas de sangre materna (19 células
fetales en 16 ml de sangre materna) mediante separación de
los mononucleares en un gradiente de densidad,
enriquecimiento de las células fetales usando anticuerpo anti
receptor de transferrina, identificación de las mismas por
medio de anticuerpos anti Hb fetal, aislamiento de glóbulos
rojos nucleados por microdisección bajo microscopía óptica y
análisis por PCR (Cheung et al, 1996). La simplificación y la
automatización parcial de este procedimiento permitirá la
introducción del diagnóstico prenatal mediante análisis de
células fetales en circulacuón materna en la práctica clínica.
DIAGNÓSTICO PRENATAL DE BETA TALASEMIA
IMB

El mayor problema con la terapia génica ha estado
relacionado con la construcción del vector. El gen
terapéutico debe ser insertado en una célula
hematopoyética y debe ser expresado a altos niveles, por
un período de tiempo extendido, de forma eritroide
específica; el vector debe ser seguro con respecto a
recombinación o mutagénesis.
Vectores lentivirales que portan un pequeño gen RNA
nuclear que codifica un RNA antisense han demostrado
que corrigen los defectos de splicing causados por las
mutaciones talasémicas (forzando la selección de sitios de
splice normal).
DIAGNÓSTICO PRENATAL DE BETA TALASEMIA
IMB

-Screening de Beta Th en 23.485 sujetos (2000-2006)
-3.934 tenían HbA2 borderline (3,1 – 3,9%).
-410 muestras (con fenotipo normal o de portador ) estudiadas por PCR por tener
parejas portadoras de beta Th clásica.
-De estos sujetos, 94 (22.9%) fueron positivos para un defecto molecular en el gen
β, ó . El defecto molecular mas prevalente fue β IVS1 nt 6, co-herencia de
mutaciones severas de β y talasemia, mutaciones en el promotor βy triplicación
de genes alfa y algunas Hbs Variantes.
-Ningún defecto molecular fue encontrado en los restantes 316 individuos. IMB

Molecular Biology in heterozygous beta Thalassemia diagnosis
XXXI World Congress of the International Society of Hematology 2007
March 20-24, 2007- Punta del Este - Uruguay
-Mujer con: GR 4,9 x 1012/l, Hb 11,7 g/dl, Hto 37%, VCM. 73,7 fL, HCM 23,4 pg.
Serie roja: microcitos hipocrómicos. Estudio de Hierro: normal.
HbA2 3,8%; Hb F 1%.
-La elevación de Hb A2 es la característica más importante en la identificación
de Th heterocigotas. Sin embargo algunos portadores pueden tener Hb A2
normal o en el límite inferior del rango para un portador.
-En 124 portadores de Th identificamos tres individuos con la mutación + I-6;
Hb A2 5,5; 5,6 y 4,9 %.
-Altay y col encontraron que el aumento de Hb A2 no se correlaciona con la
severidad de la mutación responsable de talasemia; Stefanis L y col
encontraron valores más bajos de Hb A2 en pacientes portadores de la
mutación + 1-6.
-PCR-ARMS + 1-6: positivo Bragós et al., 2007 IMB

¡¡¡MUCHAS GRACIAS!!!
IMB